We apply high-throughput, high-resolution technologies to decipher enhancer logic and map gene regulatory networks, such as single-cell RNA-seq for transcriptomics and single-cell ATAC-seq for chromatin accessibility. To test the activities of promoters and enhancers we use massively parallel enhancer-reporter assays. Our favorite model systems include Drosophila as well as human organoids and cancer cells.
We use and develop bioinformatics methods for regulatory network inference and computational modeling of enhancers, such as machine learning and advanced motif discovery. Using these, we have deciphered the enhancer code of melanoma, the fly brain, mammalian liver, mammalian and avian pallia, and others. Some of the bioinformatics methods we have developed and made available to the community include SCENIC+, i-cisTarget, and SCOPE.
We develop microfluidics chips, including droplet microfluidics for single-cell assays. We also develop microfluidic devices to analyse 3D tumoroids (organ-on-chip) and single-cell migration, in combination with lens-free imaging.
 We combine machine learning with epigenome profiling to decode enhancer logic. To test enhancers we developed a massively parallel enhancer-reporter assay, called CHEQ-seq.
Our enhancer modeling focuses on mammalian TFs, such as TP53, SOX10/SOX9, GRHL1/2/3, AP-1, and TEADs; as well as on Drosophila TFs involved in eye development (e.g. Glass, Optix, sine oculis), epithelial development (Grainyhead), and tumour development (AP-1, STAT92E, and Scalloped).
We combine machine learning with epigenome profiling to decode enhancer logic. To test enhancers we developed a massively parallel enhancer-reporter assay, called CHEQ-seq.
Our enhancer modeling focuses on mammalian TFs, such as TP53, SOX10/SOX9, GRHL1/2/3, AP-1, and TEADs; as well as on Drosophila TFs involved in eye development (e.g. Glass, Optix, sine oculis), epithelial development (Grainyhead), and tumour development (AP-1, STAT92E, and Scalloped).




By comparing transcriptomes, chromatin state and cis-regulatory modules across species, we learn about enhancer logic and the evolution of gene regulatory networks. We use RNA-seq, FAIRE-seq, and ATAC-seq across Drosophila species, alongside Ornstein-Uhlenbeck models to connect CRM evolution with variation in chromatin accessibility. We have also studied the evolution of epidermal and metabolic GRNs between Drosophila and Daphnia.

We are interested in deciphering regulatory programs of transcriptional state switches in mammalian systems, including human and mouse. To study the cis-regulatory code in mammalian genomes we mainly use cancer cells as model system. During cancer progression, gene expression profiles can change, causing regulatory heterogeneity in tumors. This heterogeneity has an important impact on therapy response, since some cell states may be more or less vulnerable to a particular drug therapy.
We study neuronal and glial cell types in the ageing Drosophila brain using single-cell RNA-seq, and compare normal cell states with disease mutations involved in Parkinson’s and Alzheimer’s disease.

 The eye-antennal disc is a classical model system to study cellular differentiation. We use this system to unravel new genomic regulatory “recipes” that control cell fate decisions, such as photoreceptor specification and differentiation.
We also perturb this system using irradiation, transcription factor perturbations, and RasV12-driven malignant transformation, to study cancer-related transcriptional changes, controlled by JNK, EGFR, and Hippo signaling pathways.
The eye-antennal disc is a classical model system to study cellular differentiation. We use this system to unravel new genomic regulatory “recipes” that control cell fate decisions, such as photoreceptor specification and differentiation.
We also perturb this system using irradiation, transcription factor perturbations, and RasV12-driven malignant transformation, to study cancer-related transcriptional changes, controlled by JNK, EGFR, and Hippo signaling pathways.

Data-driven research in our lab is powered by machine learning and artificial intelligence (AI) to help us guide and understand more about biological systems and processes. Here is a non-exhaustive list what the lab has been and is currently working on:
Single-cell transcriptomics (scRNA-seq) and single-cell epigenomics (scATAC-seq) data revolutionize the field of regulatory genomics.

 We combine new computational strategies (e.g., SCENIC, cisTopic) with state-of-the-art single-cell measurements (Drop-seq, 10X, InDrops, SeqWell) to decipher cis-regulatory “programs”, to reverse engineer gene regulatory networks, and to better define cell types and cell state transitions.
We combine new computational strategies (e.g., SCENIC, cisTopic) with state-of-the-art single-cell measurements (Drop-seq, 10X, InDrops, SeqWell) to decipher cis-regulatory “programs”, to reverse engineer gene regulatory networks, and to better define cell types and cell state transitions.
We develop new computational approaches that exploit single-cell technologies to link genome variation with changes in epigenome, transcriptome, proteome, and phenome.
We apply this to human melanoma (e.g., phenotype switching), to the mouse liver, to the developing Drosophila eye and to ageing/neurodegeneration in the Drosophila brain. See also our collaborations.

 We develop new bioinformatics tools for motif and CRM detection, and for gene regulatory network inference, such as i-cisTarget, iRegulon, and TOUCAN. We also maintain a large collection of curated position weight matrices (currently > 20.000).
We exploit single-cell RNA-seq and single-cell ATAC-seq data to improve the identification of GRNs and enhancers, with our tools SCENIC and cisTopic.
We develop new bioinformatics tools for motif and CRM detection, and for gene regulatory network inference, such as i-cisTarget, iRegulon, and TOUCAN. We also maintain a large collection of curated position weight matrices (currently > 20.000).
We exploit single-cell RNA-seq and single-cell ATAC-seq data to improve the identification of GRNs and enhancers, with our tools SCENIC and cisTopic.
![]()
SCENIC+ is a python package to build gene regulatory networks (GRNs) using combined or separate single-cell gene expression (scRNA-seq) and single-cell chromatin accessibility (scATAC-seq) data. This software is part of SCENIC Suite.
BioRXiv Preprint Github Read the Docs![]() ScoMAP (Single-Cell Omics Mapping into spatial Axes using Pseudotemporall ordering) is an R package to spatially integrate single-cell omics data into virtual cells. This software is part of SCENIC Suite.
ScoMAP (Single-Cell Omics Mapping into spatial Axes using Pseudotemporall ordering) is an R package to spatially integrate single-cell omics data into virtual cells. This software is part of SCENIC Suite.
![]() VSN-Pipelines Is a repository of pipelines for single-cell data analysis in Nextflow DSL2. It contains multiple workflows for analyzing single cell transcriptomics data, and depends on a number of tools, which are organized into submodules within the VIB-Singlecell-NF organization. This software is part of SCENIC Suite.
VSN-Pipelines Is a repository of pipelines for single-cell data analysis in Nextflow DSL2. It contains multiple workflows for analyzing single cell transcriptomics data, and depends on a number of tools, which are organized into submodules within the VIB-Singlecell-NF organization. This software is part of SCENIC Suite.
![]() SCope is a fast visualization tool for large-scale and high dimensional scRNA-seq datasets. Visit https://scope.aertslab.org to test out SCope on several published datasets! This software is part of SCENIC Suite.
SCope is a fast visualization tool for large-scale and high dimensional scRNA-seq datasets. Visit https://scope.aertslab.org to test out SCope on several published datasets! This software is part of SCENIC Suite.
![]() cisTopic is an R package to simultaneously identify cell states and cis-regulatory topics from single cell epigenomics data. This software is part of SCENIC Suite.
cisTopic is an R package to simultaneously identify cell states and cis-regulatory topics from single cell epigenomics data. This software is part of SCENIC Suite.
pySCENIC is a lightning-fast python implementation of the SCENIC pipeline (Single-CEll regulatory Network Inference and Clustering) which enables biologists to infer transcription factors, gene regulatory networks and cell types from single-cell RNA-seq data. This software is part of SCENIC Suite.
Github PyPi Read the Docs![]() Arboreto is a computational framework that offers scalable implementations of Gene Regulatory Network inference algorithms. It currently supports GRNBoost2 and GENIE3 (Huynh-Thu et al., 2010). This software is part of SCENIC Suite.
Arboreto is a computational framework that offers scalable implementations of Gene Regulatory Network inference algorithms. It currently supports GRNBoost2 and GENIE3 (Huynh-Thu et al., 2010). This software is part of SCENIC Suite.
![]() SCENIC is an R package to infer Gene Regulatory Networks and cell types from single-cell RNA-seq data. This software is part of SCENIC Suite.
SCENIC is an R package to infer Gene Regulatory Networks and cell types from single-cell RNA-seq data. This software is part of SCENIC Suite.
Spatial transcriptomics workflows using barcoded capture arrays are commonly used for resolving gene expression in tissues. However, existing techniques are either limited by capture array density or are cost prohibitive for large scale atlasing. We present Nova-ST is a dense nano-patterned spatial transcriptomics technique derived from randomly barcoded Illumina sequencing flow cells. Nova-ST enables customized, low cost, flexible, and high-resolution spatial profiling of large tissue sections. Benchmarking on mouse brain sections demonstrates significantly higher sensitivity compared to existing methods, at reduced cost.
BioRXiv Preprint Github Website
An open-source droplet microfluidics platform for single-cell RNA-seq and single-cell ATAC-seq strongly inspired by inDrop and Drop-seq. Developed at Aerts lab in close collaboration with the Single cell and Microfluidics Expertise Unit at the VIB Center for Brain and Disease Research
BioRXiv Preprint Paper Github WebsiteThis study explores enhancer codes, comparing them between mammalian neocortex and bird pallium using deep learning and transcriptomics data. We found that while non-neuronal and GABAergic cells are similar across species, excitatory neurons diverge significantly. Interestingly, some excitatory neuron enhancer codes are still shared, proposing novel cell type homologies. This research also introduces methods to compare cell types across species based on their genomic codes.
You can now register for our SCENIC+ webinar (March 26, 5:00 p.m. CET): https://forms.gle/z6fVAwaHMPtrGPuA6
Spatial transcriptomics workflows using barcoded capture arrays are commonly used for resolving gene expression in tissues. However, existing techniques are either limited by capture array density or are cost prohibitive for large scale atlasing. We present Nova-ST, a dense nano-patterned spatial transcriptomics technique derived from randomly barcoded Illumina sequencing flow cells. Nova-ST enables customized, low cost, flexible, and high-resolution spatial profiling of large tissue sections. Benchmarking on mouse brain sections demonstrates significantly higher sensitivity compared to existing methods, at reduced cost.
Our study provides a multi-modal understanding of the regulatory code underlying hepatocyte identity and their zonation state, that can be exploited to engineer enhancers with specific activity levels and zonation patterns.
We implemented and compared three different enhancer-design strategies, each built on a deep learning model: (1) directed sequence evolution; (2) directed iterative motif implanting; and (3) generative design. We evaluated the function of fully synthetic enhancers to specifically target brain cell types in Drosophila, and melanoma cell states in human. We exploited this concept further by creating “dual-code” enhancers that target two cell types, and minimal enhancers smaller than 50 base pairs that are fully functional.
https://twitter.com/ibrahimihsan/status/1552370069681864709?s=20&t=g19-Ql8vVgPwEsEQ4cUTIg
In a proof of concept study, we have applied spatial transcriptomics using a selected gene panel to pinpoint the locations of 150 mRNA species in the adult fly. This enabled us to map unknown cell types identified in the Fly Cell Atlas to their spatial locations in the brain. Additionally, spatial transcriptomics discovered interesting principles of mRNA localization in large crowded muscle cells that may spark future mechanistic investigations. Furthermore, we present a set of computational tools that will allow for easier integration of spatial transcriptomics and single-cell datasets.
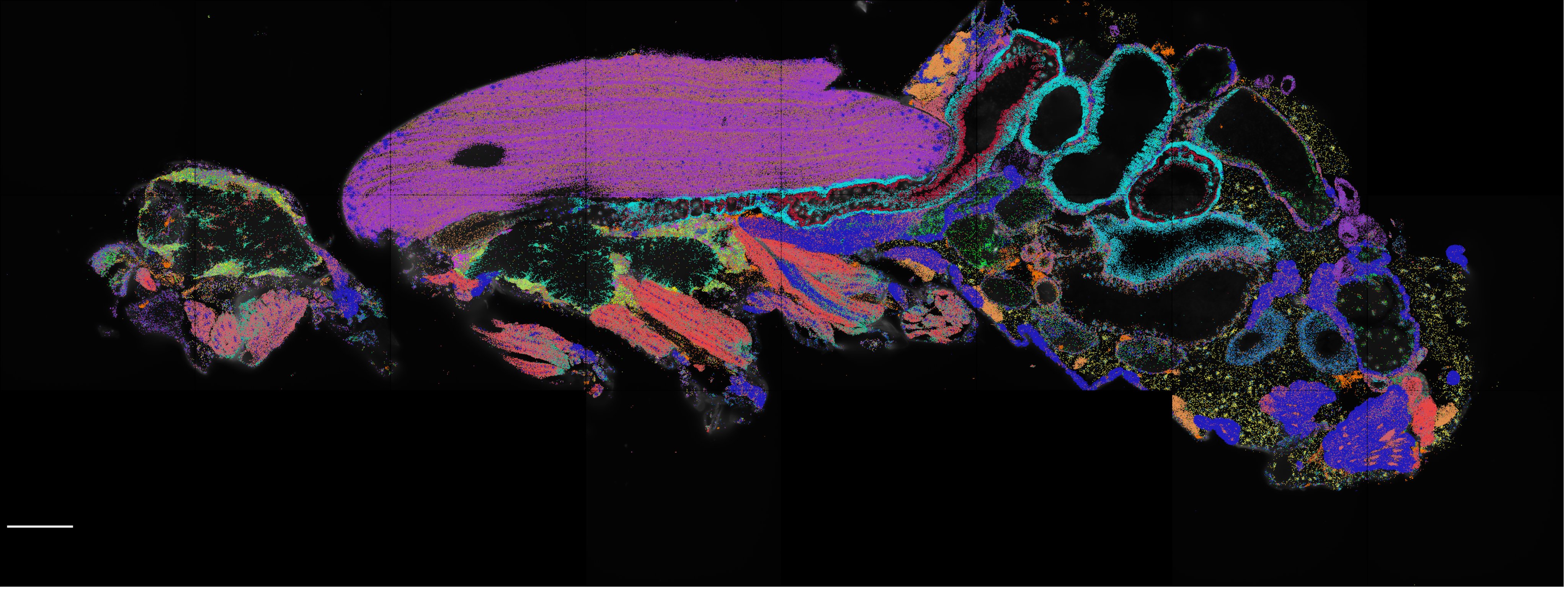
A systematic examination of eight different single-cell assay for transposase-accessible chromatin by sequencing (scATAC-seq) technologies revealed marked differences in the complexity of sequencing libraries and the specificity of DNA tagmentation that they achieve. Our pipeline for universal mapping of scATAC-seq data (PUMATAC) allowed a fair benchmarking of existing methods and enables the seamless integration of future datasets and technologies.
SCENIC+ is a new method for the inference of eGRNs. SCENIC+ predicts genomic enhancers along with candidate upstream transcription factors (TF) and links these enhancers to candidate target genes. Specific TFs for each cell type or cell state are predicted based on the concordance of TF binding site accessibility, TF expression, and target gene expression.
We characterized the regulatory states that emerge and cooperate in the wound response, using the Drosophila melanogaster wing disc as a model system, and compare these with cancer cell states induced by rasV12scrib-/- in the eye disc. We used single-cell multiome profiling to derive enhancer gene regulatory networks (eGRNs) by integrating chromatin accessibility and gene expression signals.
New computational method uses convolutional neural networks for cis-regulatory sequence analysis to analyze and cluster scATAC-seq data.
Stein Aerts receives an ERC Advanced Grant for “Genome2Cells” where we will study how the genome “translates” into cell types.
EMBO, or the European Molecular Biology Organization, brings together top researchers in the life sciences to promote collaboration and scientific progress. Each year, EMBO elects new members to join its ranks. Being elected as an EMBO member is an indication of a strong, high-quality research program that seeks to answer the molecular riddles in the life sciences. This year, Prof. Sarah-Maria Fendt (VIB-KU Leuven Center for Cancer Biology) and Prof. Stein Aerts (VIB-KU Leuven Center for Brain & Disease Research) join the other VIB EMBO members.
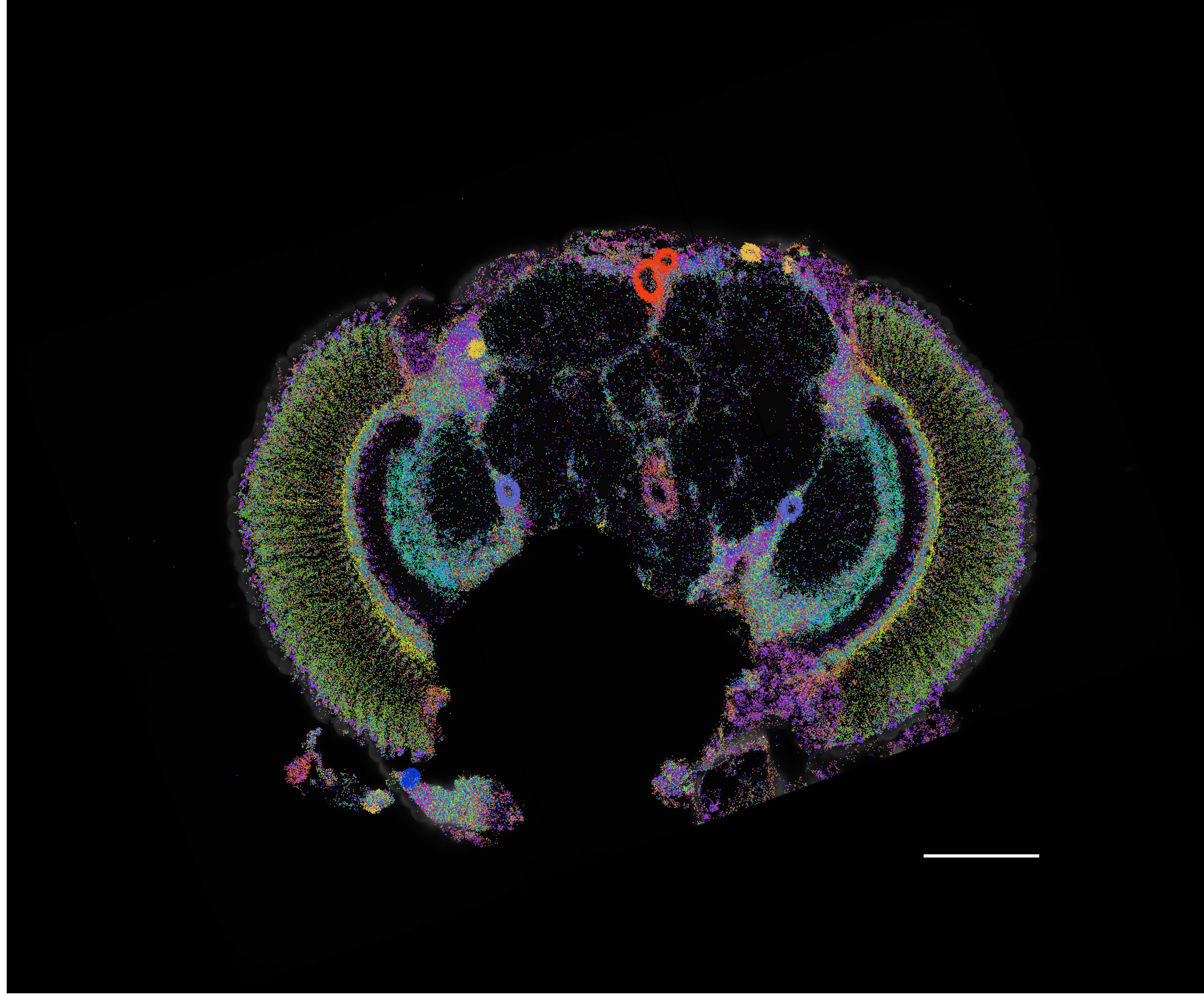
A chromatin accessibility atlas of 240,919 cells in the adult and developing Drosophila brain reveals 95,000 enhancers, that are integrated in cell-type specific enhancer gene regulatory networks and decoded into combinations of functional transcription factor binding sites using deep learning.
Multi-level massively parallel reporter assays (H3K27ac, ATAC and short tiles) in a panel of melanoma cell lines, together with a deep learning model, reveal location, multiplicity, and grammar of subtype specific enhancers.
For more than 100 years, the fruit fly Drosophila melanogaster has been one of the most studied model organisms. Here, we present a single-cell atlas of the adult fly, Tabula Drosophilae, that includes 580,000 nuclei from 15 individually dissected sexed tissues as well as the entire head and body, annotated to >250 distinct cell types. We provide an in-depth analysis of cell type–related gene signatures and transcription factor markers, as well as sexual dimorphism, across the whole animal. Analysis of common cell types between tissues, such as blood and muscle cells, reveals rare cell types and tissue-specific subtypes. This atlas provides a valuable resource for the Drosophila community and serves as a reference to study genetic perturbations and disease models at single-cell resolution.
Our lab developed HyDrop, a flexible and open-source droplet microfluidic platform for scRNA-seq and scATAC-seq experiments. We applied HyDrop-ATAC and HyDrop-RNA to flash-frozen mouse cortex and generated 7996 high-quality single-cell chromatin accessibility profiles and 9508 single-cell transcriptomes closely matching reference single-cell gene data. Additionally, we leveraged HyDrop-RNA’s high capture rate to analyze a small population of FAC-sorted neurons from the Drosophila brain, confirming the protocol’s applicability to low input samples and small cells. Our publication includes step-by-step protocols for producing dissolvable barcoded hydrogel beads and applying these beads in scRNA-seq and scATAC-seq experiments.
Genomic sequence variation within enhancers and promoters can have a significant impact on the cellular state and phenotype. Here we generate phased whole genomes with matched chromatin accessibility, histone modifications, and gene expression for 10 melanoma cell lines. We find that training a specialized deep learning model, called DeepMEL2, on melanoma chromatin accessibility data can capture the various regulatory programs of the melanocytic and mesenchymal-like melanoma cell states.

In this Primer, we discuss these biochemical methods, as well as bioinformatics tools for analysing and interpreting the generated data, and insights into the key regulators underlying developmental, evolutionary and disease processes. We outline standards for data quality, reproducibility and deposition used by the genomics community.
Melanoma cells can switch between a melanocytic and a mesenchymal-like state. Scattered evidence indicates that additional intermediate state(s) may exist. Here, to search for such states and decipher their underlying gene regulatory network (GRN), we studied 10 melanoma cultures using single-cell RNA sequencing (RNA-seq) as well as 26 additional cultures using bulk RNA-seq.
Genomic enhancers form the central nodes of gene regulatory networks by harbouring combinations of transcription factor binding sites. In order to unravel the enhancer logic of the two most common melanoma cell states, namely the melanocytic and mesenchymal-like state, we combined comparative epigenomics with machine learning. By profiling chromatin accessibility using ATAC-seq on a cohort of 27 melanoma cell lines across six different species, we demonstrate the conservation of the two main melanoma states and their underlying master regulators.
Single‐cell technologies allow measuring chromatin accessibility and gene expression in each cell, but jointly utilizing both layers to map bona fide gene regulatory networks and enhancers remains challenging. Here, we generate independent single‐cell RNA ‐seq and single‐cell ATAC ‐seq atlases of the Drosophila eye‐antennal disc and spatially integrate the data into a virtual latent space that mimics the organization of the 2D tissue using ScoMAP (Single‐Cell Omics Mapping into spatial Axes using Pseudotime ordering).

This protocol explains how to perform a fast SCENIC analysis alongside standard best practices steps on single-cell RNA-sequencing data using software containers and Nextflow pipelines.
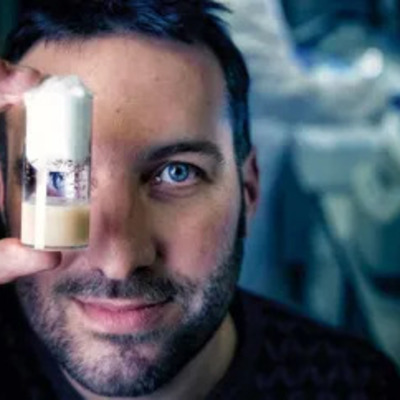
“It’s still me,” says Stein Aerts about his diverse science chapters to date. […] Click on link below to read more.
Single-cell epigenomics provides new opportunities to decipher genomic regulatory programs from heterogeneous samples and dynamic processes. We present a probabilistic framework called cisTopic, to simultaneously discover “cis-regulatory topics” and stable cell states from sparse single-cell epigenomics data.
A single-cell atlas of the adult fly brain during aging:
Using ATAC-seq across a panel of Drosophila inbred strains, we found that SNPs affecting binding sites of the TF Grainy head (Grh) causally determine the accessibility of epithelial enhancers.
The Fly Cell Atlas will bring together Drosophila researchers interested in single-cell genomics, transcriptomics, and epigenomics, to build comprehensive cell atlases during different developmental stages and disease models.
Here we show that the Hippo pathway is critical for this decision. Loss of Hippo switches Ras activation from promoting cellular differentiation to aggressive cellular proliferation.
Using two complementary techniques of multiplex enhancer-reporter assays, we discovered that functional enhancers could be discriminated from nonfunctional binding events by the occurrence of a single TP53 canonical motif.
Using regulatory landscapes and in silico analysis, we show that transcriptional reprogramming underlies the distinct cellular states present in melanoma. Furthermore, it reveals an essential role for the TEADs, linking it to clinically relevant mechanisms such as invasion and resistance.
![]()
Together with B. Deplancke and R. Zinzen we founded the Fly Cell Atlas.
The Fly Cell Atlas will bring together Drosophila researchers interested in single-cell genomics, transcriptomics, and epigenomics, to build comprehensive cell atlases during different developmental stages and disease models.
Go to flycellatlas.org MendelCraft is a MineCraft mod developed in the lab to teach children about DNA, genetics, and the laws of Mendel.
You can visit the website at https://mendelcraft.aertslab.org/
MendelCraft is a MineCraft mod developed in the lab to teach children about DNA, genetics, and the laws of Mendel.
You can visit the website at https://mendelcraft.aertslab.org/
ERC funded postdoc: (deep) learning the genomic regulatory code: https://jobs.vib.be/j/49442/

PhD student: deep learning for cancer genomics: https://jobs.vib.be/j/49440/









Stein Aerts |
Herestraat 49, PO Box 602, 3000 LEUVEN, Belgium |
+32-16-33 07 10 |


















































{kind=link}
{kind=link}
{kind=link}